计算干货 | 基于理论与计算化学Gaussian软件研究缓蚀剂的缓蚀机理
2017-08-30 12:23:48
作者:本网整理 来源:材料人
(1)基于Gaussian软件研究缓蚀剂
为了研究不同缓蚀剂的缓蚀机理,我采用了Gaussian软件,在6-31G++(d,p)基组水平上,对所研究的缓蚀剂苯并三唑和硫脲分子进行了几何构型优化,并进行频率分析,确保所得结构均为势能面上的极小点无虚频。
通过使用ChemOffice软件中的Chemdraw软件,绘制了两种缓蚀剂苯并三唑和硫脲分子的结构式,如图27所示。
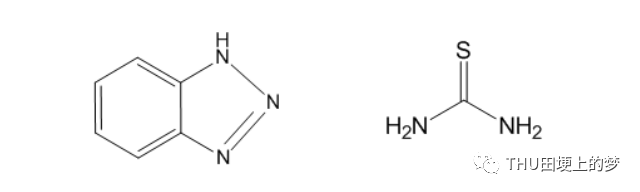
图27. 两种缓蚀剂苯并三唑(左)和硫脲(右)分子的结构式
在同一基组水平上,分别计算了苯并三唑和硫脲缓蚀剂分子的前线轨道分布和Fukui指数,用于分析分子的反应活性区域和吸附位点。
获得相关参数:最高占据轨道HOMO和最低空轨道LUMO的能量及两者之间的能量差,全局参量,如电离能、电子亲和能、电负性、硬度、偶极距、总能量、分子体积和电子分布等参量。
缓蚀剂分子的电负性和硬度的关系式如下:
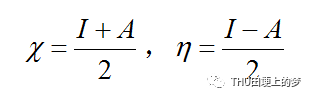
从缓蚀剂分子中转移到金属的电子数是根据从所获得的和的值计算得到的,计算公式如下:
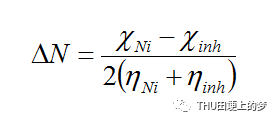
其中使用理论值,由于它比中性原子更加柔软,因此,通过假设金属的硬度为一个整体,即I=A。
(2)缓蚀剂分子的优化几何构型
密度泛函理论DFT是一种经济、高效的量子化学计算方法,它可以提供足够精确的分子几何构型和电子分布等信息。
首先我对这两种缓蚀剂苯并三唑和硫脲分子进行分子结构优化。图28是硫脲分子的最优构型。所涉及的量子化学参数均是在分子最优构型的基础上计算所得。
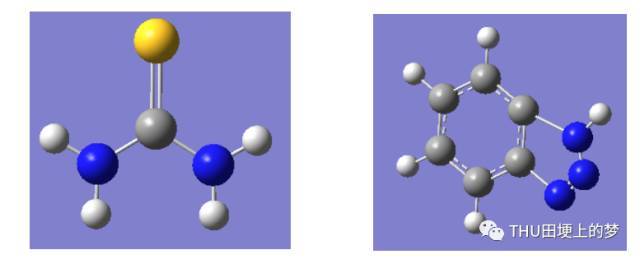
图28. 硫脲分子(左)和苯并三唑分子(右)的最优构型
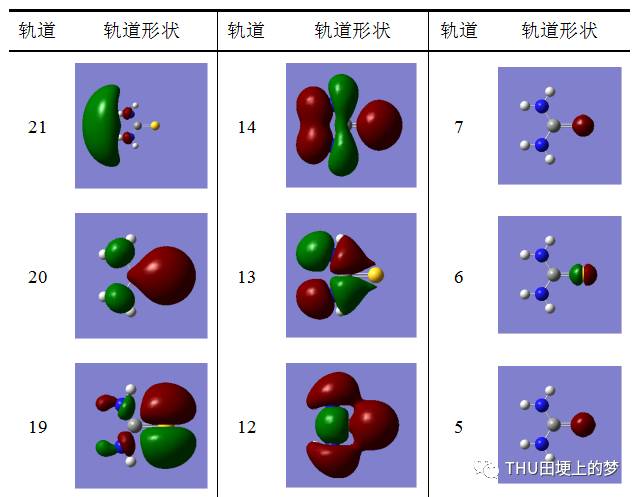
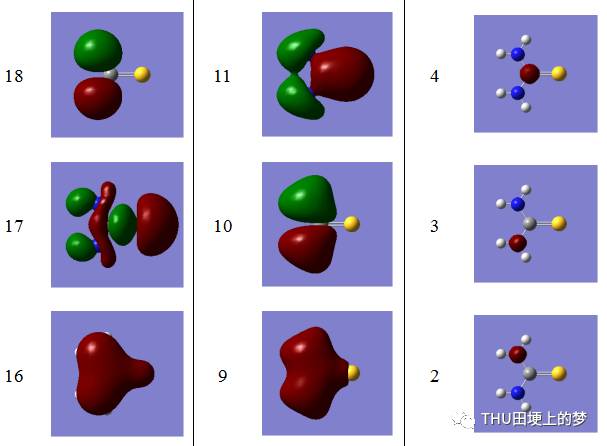
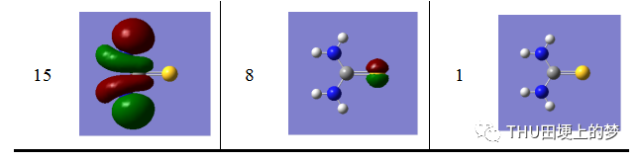
表13是使用Gaussian软件计算得到的硫脲分子的分子轨道数目和形状。
表13. 硫脲分子的分子轨道形状
(3)缓蚀剂全局反应活性
我对所研究的缓蚀剂分子做了模拟计算,并得出相应的量子化学参数,可以用于分析缓蚀剂分子的反应活性和选择性能。
量子化学的前线轨道理论认为反应物间的相互作用仅发生在分子的前线轨道之间,分子中的EHOMO是分子给电子能力的大小,分子的EHOMO越高,则该分子越易提供电子参与亲核反应。分子的EHOMO是与分子的电子亲和能有关,其值越低,该分子则有较强的接受电子的能力。因此分子的最高占据轨道HOMO和最低空轨道LUMO是分析缓蚀剂在金属表面吸附行为的重要依据。
图为无溶剂状态下两种缓蚀剂苯并三唑和硫脲分子的HOMO和LUMO分布图。
表14是两种缓蚀剂苯并三唑和硫脲分子量化计算的结构参数。
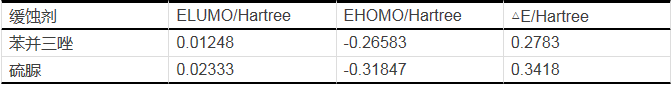
从表14中可以看出,从EHOMO的大小来看,苯并三唑的EHOMO为-0.26583 Hartree,硫脲的EHOMO为-0.31847 Hartree,EHOMO越大越好,说明苯并三唑的缓蚀效率最高,与极化曲线得出的结论相一致。
从ELUMO来看,苯并三唑的ELUMO为0.01248 Hartree,硫脲的ELUMO为0.02333 Hartree,ELUMO越小越好,苯并三唑的缓蚀效率最好。再从△E来看,苯并三唑的△E为0.2783 Hartree,硫脲的△E为0.3418 Hartree,△E越小越好,苯并三唑的缓蚀效率最好。从这三组参数的结果中我们可以确定出苯并三唑缓蚀剂效果比硫脲缓蚀剂效果好。
(4)结论
1. 苯并三唑的缓蚀效率优于硫脲的缓蚀效率,其中,苯并三唑属于以抑制阳极为主的混合型缓蚀剂,硫脲属于以抑制阴极为主的混合型缓蚀剂。
2. 两种缓蚀剂苯并三唑和硫脲分子在铝和镍表面的吸附均服从Langmuir等温式,吸附过程是放热过程,属于物理吸附。因此理论上,在所研究的温度范围内两种缓蚀剂苯并三唑和硫脲分子的缓蚀性能随温度的升高而降低。
3. 运用量子化学理论和分子设计等先进科学技术在理论上预测和计算缓蚀剂,在特定环境中的缓蚀效率和缓蚀机理等。在某种程度上,可以减少实验时测试的工作量,有利于合理选择缓蚀剂,克服实验的盲目性。
更多关于材料方面、材料腐蚀控制、材料科普等方面的国内外最新动态,我们网站会不断更新。希望大家一直关注国家材料腐蚀与防护科学数据中心http://www.ecorr.org
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。