分子振动也可能引起分子极化率的变化,产生拉曼光谱。拉曼光谱不是观察光的吸收, 而是观察光的非弹性散射。非弹性散射光很弱,过去较难观测。激光拉曼光谱的出现使灵敏度和分辨力大大提高,应用日益广泛。
拉曼散射效应的进展
1928年,印度物理学家拉曼(C.V.Raman)首次发现曼散射效应,荣获1930年的诺贝尔物理学奖。
1928-1940年,拉曼光谱成为研究分子结构的主要手段。
1960年以后,激光技术的发展使拉曼技术得以复兴。由于激光束的高亮度、方向性和偏振性等优点,成为拉曼光谱的理想光源。随探测技术的改进和对被测样品要求的降低,目前在物理、化学、医药、工业等各个领域拉曼光谱得到了广泛的应用,越来越受研究者的重视。
什么是拉曼光谱分析法
拉曼光谱分析法是基于印度科学家C.V.拉曼(Raman)所发现的拉曼散射效应,对与入射光频率不同的散射光谱进行分析以得到分子振动、转动方面信息,并应用于分子结构研究的一种分析方法。
拉曼光谱仪原理
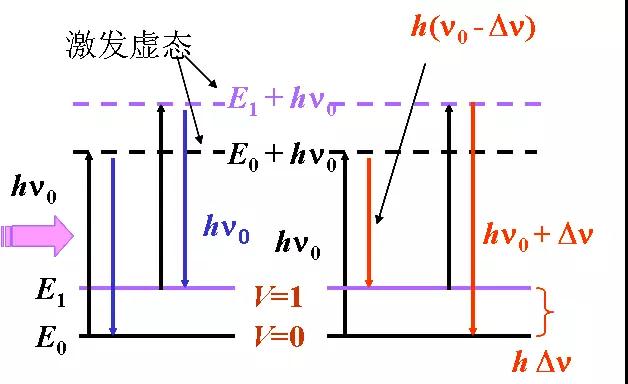
当光线照射到分子并且和分子中的电子云及分子键结产生相互作用,就会发生拉曼效应。对于自发拉曼效应,光子将分子从基态激发到一个虚拟的能量状态。当激发态的分子放出一个光子后并返回到一个不同于基态的旋转或振动状态。在基态与新状态间的能量差会使得释放光子的频率与激发光线的波长不同。
如果最终振动状态的分子比初始状态时能量高,所激发出来的光子频率则较低,以确保系统的总能量守衡。这一个频率的改变被名为Stokes shift。如果最终振动状态的分子比初始状态时能量低,所激发出来的光子频率则较高,这一个频率的改变被名为Anti-Stokes shift。拉曼散射是由于能量透过光子和分子之间的相互作用而传递,就是一个非弹性散射的例子。
关于振动的配位,分子极化电位的改变或称电子云的改变量,是分子拉曼效应必定的结果。极化率的变化量将决定拉曼散射强度。该模式频率的改变是由样品的旋转和振动状态决定。
1.Rayleigh散射:弹性碰撞;无能量交换,仅改变方向;
2.Raman散射:非弹性碰撞;方向改变且有能量交换;
拉曼光谱的特征
1. 对不同物质Raman 位移不同;
2.对同一物质Δν与入射光频率无关;是表征分子振-转能级的特征物理量;是定性与结构分析的依据;
3.拉曼线对称地发布在瑞利线两侧,长波一侧为斯托克斯线,短波一侧为反斯托克斯线;
4.斯托克斯线强度比反斯托克斯线强;
拉曼谱图的构成和特征一张拉曼谱图通常由一定数量的拉曼峰构成,每个拉曼峰代表了相应的拉曼位移和强度。每个谱峰对应于一种特定的分子键振动,其中既包括单一的化学键,例如C-C,C=C,N-O,C-H等,也包括由数个化学键组成的基团的振动,例如苯环的呼吸振动、多聚物长链的振动以及晶格振动等。
拉曼光谱可以提供样品化学结构、相和形态、结晶度及分子相互作用的详细信息。

主要的拉曼光谱仪
激光Raman光谱仪(laser Raman spectroscopy)
Ar激光器:
波长: 514.5nm,488.0nm;
单色器:
光栅,多单色器;
检测器:
光电倍增管,光子计数器;

傅立叶变换-拉曼光谱仪(FT-Raman spectroscopy)光源:Nd-YAG钇铝石榴石激光器(1.064um);检测器:高灵敏度的铟镓砷探头;
特点:
(1)避免了荧光干扰;(2)精度高;(3)消除了瑞利谱线;(4)测量速度快。
拉曼光谱的分析方向
拉曼光谱仪分析技术是以拉曼效应为基础建立起来的分子结构表征技术,其信号来源与分子的振动和转动。
拉曼光谱的分析方向有:
定性分析:不同的物质具有不同的特征光谱,因此可以通过光谱进行定性分析。
结构分析:对光谱谱带的分析,又是进行物质结构分析的基础。
定量分析:根据物质对光谱的吸光度的特点,可以对物质的量有很好的分析能力。
拉曼光谱的应用
由拉曼光谱可以获得有机化合物的各种结构信息:
1 同种分子的非极性键S-S,C=C,N=N,C ≡C产生强拉曼谱带, 随单键到双键再到三键谱带强度增加。
2 红外光谱中,由C ≡N,C=S,S-H伸缩振动产生的谱带一般较弱或强度可变,而在拉曼光谱中则是强谱带。
3 环状化合物的对称呼吸振动常常是最强的拉曼谱带。
4.在拉曼光谱中,X=Y=Z,C=N=C,O=C=O-这类键的对称伸缩振动是强谱带,反这类键的对称伸缩振动是弱谱带。红外光谱与此相反。
5 C-C伸缩振动在拉曼光谱中是强谱带。
6 醇和烷烃的拉曼光谱是相似的:I. C-O键与C-C键的力常数或键的强度没有很大差别。II. 羟基和甲基的质量仅相差2单位。 III.与C-H和N-H谱带比较,O-H拉曼谱带较弱。
拉曼光谱仪用于分析的优、缺点
1.拉曼光谱用于分析的优点
拉曼光谱的分析方法不需要对样品进行前处理,也没有样品的制备过程,避免了一些误差的产生,并且在分析过程中操作简便,测定时间短,灵敏度高等优点
2.拉曼光谱用于分析的不足
(1)拉曼散射面积(2)不同振动峰重叠和拉曼散射强度容易受光学系统参数等因素的影响(3)荧光现象对傅立叶变换拉曼光谱分析的干扰(4)在进行傅立叶变换光谱分析时,常出现曲线的非线性的问题(5)任何一物质的引入都会对被测体体系带来某种程度的污染,这等于引入了一些误差的可能性,会对分析的结果产生一定的影响。
拉曼光谱经典问题集锦
一、如何用拉曼光谱仪测透明的有机物液体,测试时放到了玻璃片上测出来的结果是玻璃的光谱。
1.我今天还在用激光拉曼测聚苯乙烯,没有出现你说的情况啊是不是玻璃管被污染的厉害?
2.你测出的玻璃的信号,有没有可能们焦点位置不对?
3.应该是聚焦位置不对,聚在玻璃上了,我以前也犯过同样的错误。
4.用凹面载玻片,液体量会比较多,然后用显微镜聚焦好就可以了,如果,液体有挥发性,最好液体上用盖玻片,然后,焦点聚焦到盖玻片以下。
※如果还不行,你可以查一下“液芯光纤”这个东东
5.建议:
(1)有机液体里面的分析物质浓度多大? Raman测定的是散射光,所以在溶液中的强度相对比较底,故分析物浓度要大些。
(2)你用的是共聚焦Raman吗?聚焦点要在毛细管的溶液里面才好。可以在溶液中放点“杂物”方便聚焦。
(3)玻璃是无定形态物质,应该Raman信号比较弱才对。
二、我们这里有做生物样品的拉曼光谱的,在获得的图里面有很强的荧光,有的说,如果拉曼得不到就用其荧光谱。可我想问一下,在拉曼谱里面得到的荧光背景,是真正的荧光特征谱吗?这和荧光光谱仪里面的荧光图有什么区别?
1.原则上说,拉曼谱中的荧光和荧光谱中的荧光是一样的,只要激发波长和功率密度相同。注意横坐标要从波数变换为纳米,即用10000000nm(1cm)除以波数就行了。但有一点要注意,不同波长的激发光照射样品,得到的拉曼相近,但荧光可以有很大不同,甚至相同波长不同功率激发,荧光谱都大不一样。
2.“注意横坐标要从波数变换为纳米,即,用10000000nm(1cm)除以波数就行了”?
Raman测定的是散射光,得到的是Raman shift.Raman shift和绝对波长(荧光光谱)之间要一个转换的吧。
3.生物样品一般荧光峰比较宽,用荧光光测试之前一般先会做仪器本身曲线校正也就是仪器本身的响应曲线,这样测出的荧光峰才比较准,特别是对于宽峰更要做这个较准。
而Raman光谱一般采集的区域比较窄(指的是波长区域),一般在窄的波长范围变化不大,因此一般不考虑仪器本身响应曲线误差,但是Raman光谱来测宽荧光峰,影响就比较大。
三、什么是共焦显微拉曼光谱仪?
1.共焦拉曼指的是空间滤波的能力和控制被分析样品的体积的能力。通常主要是利用显微镜系统来实现的。
仅仅是增加一个显微镜到拉曼光谱仪上不会起到控制被测样品体积的作用的—为达到这个目的需要一个空间滤波器。
2.显微是利用了显微镜,可以观测并测量微量样品,最小1微米左右;(2)共焦是样品在显微镜的焦平面上,而样品的光谱信息被聚焦到CCD上,都是焦点,所以叫共聚焦。
3.拉曼仪器的共焦有2种呢,一种是针孔共焦,一种是赝共焦。我觉得好像不应该称为赝共焦,共聚焦有真正的定义说一定要针孔才是共聚焦吗?好像没有,顶多称为传统共聚焦或者针孔共聚焦、简单共聚焦之类的。(个人想法,大家指正。)
四、请问,测固体粉末的拉曼图谱时,对于荧光很强的物质,应该如何处理?特别是当荧光将拉曼峰湮灭时,应该怎么办?增加照射时间的方法,我试过,连续照射了4小时,结果还是有很强的荧光。我只有一台532nm的激光器,所以更换激光波长的方法目前我不能用。想问问各位,还有别的方法吗?
1.使用SERS技术或者使用很少量的样品进行测量,或者稀释你的样品到一些别的基体里面去,比如,KBr。
2.波长不可调的话,激光强度应该是可调的,你把激光强度调低点试试。这个在光源和软件上都有调的。全调到比较低的,然后再用长时间试试。
3.可以尝试找一种溶剂溶解粉末,看能不能猝灭荧光背景。采用反斯托克斯,滤光片用Nortch滤光片。
五、请问用激光拉曼仪能测量薄膜的厚度、折射率及应力吗?它能对薄膜进行那些方面的测量呢?
1.应该不能测薄膜的厚度、折射率及应力吧;2.现在的共焦显微拉曼可以做膜及不同层膜的,你的问题我觉得用椭偏仪更好;3.拉曼光谱可以测量应力,厚度好像不行;4.应力可以测,应力有差别的时候拉曼会有微小频移,其他两种没听说过拉曼能测。
六、拉曼做金属氧化物含量的下限是多少? 我有一几种氧化物的混合物,其中MoO3含量只有5%,XRD检测不到,拉曼可以吗?
应该和待测样品的拉曼活性有关,并不能绝对说一定能测到多少检测线,有些氧化物可能纯的样品也测不出光谱,信号强的则可能会低一些。
七、小弟是刚涉足拉曼这个领域,主打生物医学方面。实验中,发现温度不同时,拉曼好像也不一样。不知到哪位能帮忙解释一下这个现象?
温度升高,拉曼线会频移,线宽会变宽,只要物质状态不变,特征峰不会有太大变化,除非高温造成化学反应或者其他变化。
八、文献上说,拉曼的峰强与物质的浓度是成正比关系,那么比如我配置1mol/L的某溶液,和0.5mol/L的溶液,其峰强度是正好一半的关系吗?应用拉曼,是否能采用峰积分,或者用近红外那样的多元统计的办法来定量吗?准确度怎么样?
存在激发效率的问题,拉曼一直以来被认为只能做半定量的研究,就是因为不是线性的,有这方面的文献,具体记不清了。
九、拉曼峰1640对应的是什么东西啊?无机的……
1.这个峰一般来说是C=O双键的峰,可是你说是无机物,很有可能是某一个基团的倍频峰,看看820左右或者是某两个峰的叠加。
2.也有可能是你在测量过程当中由于激光引起的碳化物质。还有一种可能就是C=C。
3.拉曼在1610-1680波数区间有C=N双键的强吸收。
十、1.红外分析气体需要多高的分辨率? 2.拉曼光谱仪是否可分析纯金属? 3.红外与拉曼联用,BRUKER和NICOLET哪个好些?
1.分析气体时理论上最高只需0.5cm-1。实际应用上绝大部分情况下4cm-1已足够。对于气体,还是希望分辨率高一些好,一般都用1cm-1一下,这样对气体的一些微小峰的变化检测更好
2.基本上不可能:
金属不太可能作出来,一般不发生分子极化率改变。
3.这两家公司的红外各有千秋相差不多,关键是你更看重哪些指标。