
国家材料腐蚀与防护科学数据中心
National Materials Corrosion and Protection Scientific Data Center
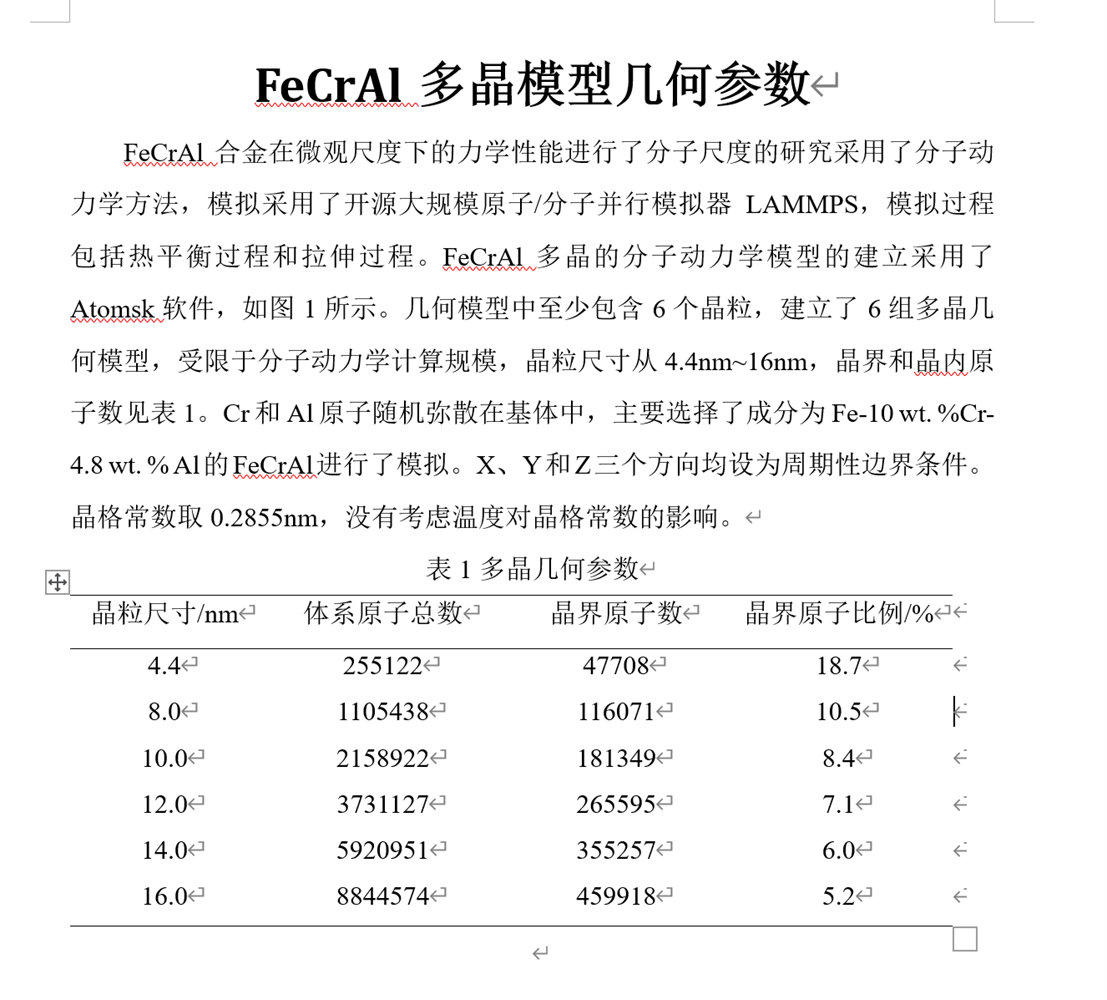
采用了开源大规模原子/分子并行模拟器LAMMPS,模拟过程包括热平衡过程和拉伸过程。FeCrAl多晶的分子动力学模型的建立采用了Atomsk软件,几何模型中至少包含6个晶粒,建立了6组多晶几何模型.
Using the open-source large-scale atomic/molecular parallel simulator LAMMPS, the simulation process includes thermal equilibrium process and tensile process. The molecular dynamics model of FeCrAl polycrystalline was established using Atomsk software, and at least 6 grains were included in the geometric model, and 6 sets of polycrystalline geometric models were established.
 国家材料腐蚀与防护科学数据中心 |
 国家高能物理科学数据中心 |
 国家基因组科学数据中心 |
 国家微生物科学数据中心 |
 国家空间科学数据中心 |
 国家天文科学数据中心 |
 国家对地观测科学数据中心 |
 国家极地科学数据中心 |
 国家青藏高原科学数据中心 |
 国家生态科学数据中心 |
 国家冰川冻土沙漠科学数据中心 |
 国家计量科学数据中心 |
 国家地球系统科学数据中心 |
 国家人口健康科学数据中心 |
 国家基础学科公共科学数据中心 |
 国家农业科学数据中心 |
 国家林业和草原科学数据中心 |
 国家气象科学数据中心 |
 国家地震科学数据中心 |
 国家海洋科学数据中心 |